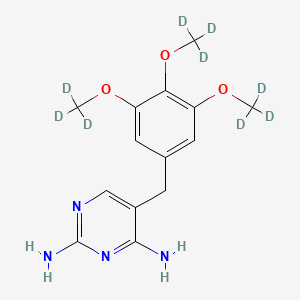
Trimethoprim-d9 (Major)
Katalognummer B562225
Molekulargewicht: 299.378
InChI-Schlüssel: IEDVJHCEMCRBQM-GQALSZNTSA-N
Achtung: Nur für Forschungszwecke. Nicht für den menschlichen oder tierärztlichen Gebrauch.
Patent
US04144263
Procedure details


A solution of α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile (7.9 g, 0.02 mol) and an equivalent amount of potassium hydroxide in ethanol (50 ml) was heated at reflux for one hour. A solution of guanidine (0.07 mol) in ethanol (50 ml) was added, and reflux was resumed. Some ethanol was allowed to boil off bringing the reaction temperature up to 85°. After about 20 hours at reflux the mixture was allowed to cool, and the product was filtered and washed with ethanol. The crude product was purified by treating with hot aqueous acetic acid and reprecipitation with ammonium hydroxide. The yield of purified trimethoprim (m.p. 197°-198°) was 3.6g (62%), its identity being confirmed by an NMR spectrum.
Name
α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile
Quantity
7.9 g
Type
reactant
Reaction Step One


Identifiers
|
REACTION_CXSMILES
|
C([C:6]([CH:22](OCC)OCC)([CH2:9][C:10]1[CH:15]=[C:14]([O:16][CH3:17])[C:13]([O:18][CH3:19])=[C:12]([O:20][CH3:21])[CH:11]=1)[C:7]#[N:8])(OCC)=O.[OH-].[K+].[NH2:31][C:32]([NH2:34])=[NH:33]>C(O)C>[NH2:34][C:32]1[N:33]=[C:7]([NH2:8])[C:6]([CH2:9][C:10]2[CH:15]=[C:14]([O:16][CH3:17])[C:13]([O:18][CH3:19])=[C:12]([O:20][CH3:21])[CH:11]=2)=[CH:22][N:31]=1 |f:1.2|
|
Inputs
Step One
|
Name
|
α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile
|
|
Quantity
|
7.9 g
|
|
Type
|
reactant
|
|
Smiles
|
C(=O)(OCC)C(C#N)(CC1=CC(=C(C(=C1)OC)OC)OC)C(OCC)OCC
|
|
Name
|
|
|
Quantity
|
0 (± 1) mol
|
|
Type
|
reactant
|
|
Smiles
|
[OH-].[K+]
|
|
Name
|
|
|
Quantity
|
50 mL
|
|
Type
|
solvent
|
|
Smiles
|
C(C)O
|
Step Two
Step Three
|
Name
|
|
|
Quantity
|
0 (± 1) mol
|
|
Type
|
solvent
|
|
Smiles
|
C(C)O
|
Conditions
Other
|
Conditions are dynamic
|
1
|
|
Details
|
See reaction.notes.procedure_details.
|
Workups
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
at reflux for one hour
|
|
Duration
|
1 h
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
reflux
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
up to 85°
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
After about 20 hours at reflux the mixture
|
|
Duration
|
20 h
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
to cool
|
FILTRATION
|
Type
|
FILTRATION
|
|
Details
|
the product was filtered
|
WASH
|
Type
|
WASH
|
|
Details
|
washed with ethanol
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
The crude product was purified
|
ADDITION
|
Type
|
ADDITION
|
|
Details
|
by treating with hot aqueous acetic acid and reprecipitation with ammonium hydroxide
|
Outcomes
Product
|
Name
|
|
|
Type
|
|
|
Smiles
|
NC1=NC=C(C(=N1)N)CC1=CC(=C(C(=C1)OC)OC)OC
|
Source
|
Source
|
Open Reaction Database (ORD) |
|
Description
|
The Open Reaction Database (ORD) is an open-access schema and infrastructure for structuring and sharing organic reaction data, including a centralized data repository. The ORD schema supports conventional and emerging technologies, from benchtop reactions to automated high-throughput experiments and flow chemistry. Our vision is that a consistent data representation and infrastructure to support data sharing will enable downstream applications that will greatly improve the state of the art with respect to computer-aided synthesis planning, reaction prediction, and other predictive chemistry tasks. |
Patent
US04144263
Procedure details
A solution of α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile (7.9 g, 0.02 mol) and an equivalent amount of potassium hydroxide in ethanol (50 ml) was heated at reflux for one hour. A solution of guanidine (0.07 mol) in ethanol (50 ml) was added, and reflux was resumed. Some ethanol was allowed to boil off bringing the reaction temperature up to 85°. After about 20 hours at reflux the mixture was allowed to cool, and the product was filtered and washed with ethanol. The crude product was purified by treating with hot aqueous acetic acid and reprecipitation with ammonium hydroxide. The yield of purified trimethoprim (m.p. 197°-198°) was 3.6g (62%), its identity being confirmed by an NMR spectrum.
Name
α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile
Quantity
7.9 g
Type
reactant
Reaction Step One
Identifiers
|
REACTION_CXSMILES
|
C([C:6]([CH:22](OCC)OCC)([CH2:9][C:10]1[CH:15]=[C:14]([O:16][CH3:17])[C:13]([O:18][CH3:19])=[C:12]([O:20][CH3:21])[CH:11]=1)[C:7]#[N:8])(OCC)=O.[OH-].[K+].[NH2:31][C:32]([NH2:34])=[NH:33]>C(O)C>[NH2:34][C:32]1[N:33]=[C:7]([NH2:8])[C:6]([CH2:9][C:10]2[CH:15]=[C:14]([O:16][CH3:17])[C:13]([O:18][CH3:19])=[C:12]([O:20][CH3:21])[CH:11]=2)=[CH:22][N:31]=1 |f:1.2|
|
Inputs
Step One
|
Name
|
α-carbethoxy-α-diethoxymethyl-β-(3,4,5-trimethoxyphenyl)propionitrile
|
|
Quantity
|
7.9 g
|
|
Type
|
reactant
|
|
Smiles
|
C(=O)(OCC)C(C#N)(CC1=CC(=C(C(=C1)OC)OC)OC)C(OCC)OCC
|
|
Name
|
|
|
Quantity
|
0 (± 1) mol
|
|
Type
|
reactant
|
|
Smiles
|
[OH-].[K+]
|
|
Name
|
|
|
Quantity
|
50 mL
|
|
Type
|
solvent
|
|
Smiles
|
C(C)O
|
Step Two
Step Three
|
Name
|
|
|
Quantity
|
0 (± 1) mol
|
|
Type
|
solvent
|
|
Smiles
|
C(C)O
|
Conditions
Other
|
Conditions are dynamic
|
1
|
|
Details
|
See reaction.notes.procedure_details.
|
Workups
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
at reflux for one hour
|
|
Duration
|
1 h
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
reflux
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
up to 85°
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
After about 20 hours at reflux the mixture
|
|
Duration
|
20 h
|
TEMPERATURE
|
Type
|
TEMPERATURE
|
|
Details
|
to cool
|
FILTRATION
|
Type
|
FILTRATION
|
|
Details
|
the product was filtered
|
WASH
|
Type
|
WASH
|
|
Details
|
washed with ethanol
|
CUSTOM
|
Type
|
CUSTOM
|
|
Details
|
The crude product was purified
|
ADDITION
|
Type
|
ADDITION
|
|
Details
|
by treating with hot aqueous acetic acid and reprecipitation with ammonium hydroxide
|
Outcomes
Product
|
Name
|
|
|
Type
|
|
|
Smiles
|
NC1=NC=C(C(=N1)N)CC1=CC(=C(C(=C1)OC)OC)OC
|
Source
|
Source
|
Open Reaction Database (ORD) |
|
Description
|
The Open Reaction Database (ORD) is an open-access schema and infrastructure for structuring and sharing organic reaction data, including a centralized data repository. The ORD schema supports conventional and emerging technologies, from benchtop reactions to automated high-throughput experiments and flow chemistry. Our vision is that a consistent data representation and infrastructure to support data sharing will enable downstream applications that will greatly improve the state of the art with respect to computer-aided synthesis planning, reaction prediction, and other predictive chemistry tasks. |
